Actualidad informática
Noticias y novedades sobre informática
La guía del fabricante de medicamentos para la galaxia
En 2016, la empresa farmacéutica Sunovion asignó a un grupo de profesionales experimentados una misión inusual. En la sede central de la firma en Marlborough, Massachusetts, a todos los químicos se les pidió que participaran en un juego para ver quién podía descubrir las mejores pistas de medicamentos nuevos. En sus puestos de trabajo había una cuadrícula de cientos de estructuras químicas, de las cuales sólo diez estaban etiquetadas con información sobre sus efectos biológicos. Los expertos tuvieron que seleccionar otras moléculas que podrían resultar ser candidatas a medicamentos, utilizando sus conocimientos de estructura química y biología, que tanto les costó aprender. De los 11 jugadores, 10 lucharon durante varias horas. Pero uno de ellos consiguió pasar en milisegundos, porque era un algoritmo.
Ese programa de computación fue la creación de Willem van Hoorn, jefe de quimioinformática de Exscientia, una empresa que usa inteligencia artificial (IA) para diseñar medicamentos. La firma, con sede en Dundee, Reino Unido, quería ampliar una incipiente asociación con Sunovion, por lo que las apuestas eran altas. «Mi credibilidad estaba en juego», dice van Hoorn. Veinte rondas de juego más tarde, él anotó los puntos. El alivio lo invadió. Su algoritmo había dominado al menos algunas de las artes oscuras de la química; sólo un experto en caza de drogas había derrotado a la máquina.
Desde entonces, Exscientia y Sunovion han seguido trabajando juntas para descubrir fármacos psiquiátricos. «Esta competencia realmente ayudó a que la gente que toma las decisiones de investigación química se comprometiera», dice Scott Brown, director de química computacional de Sunovion.
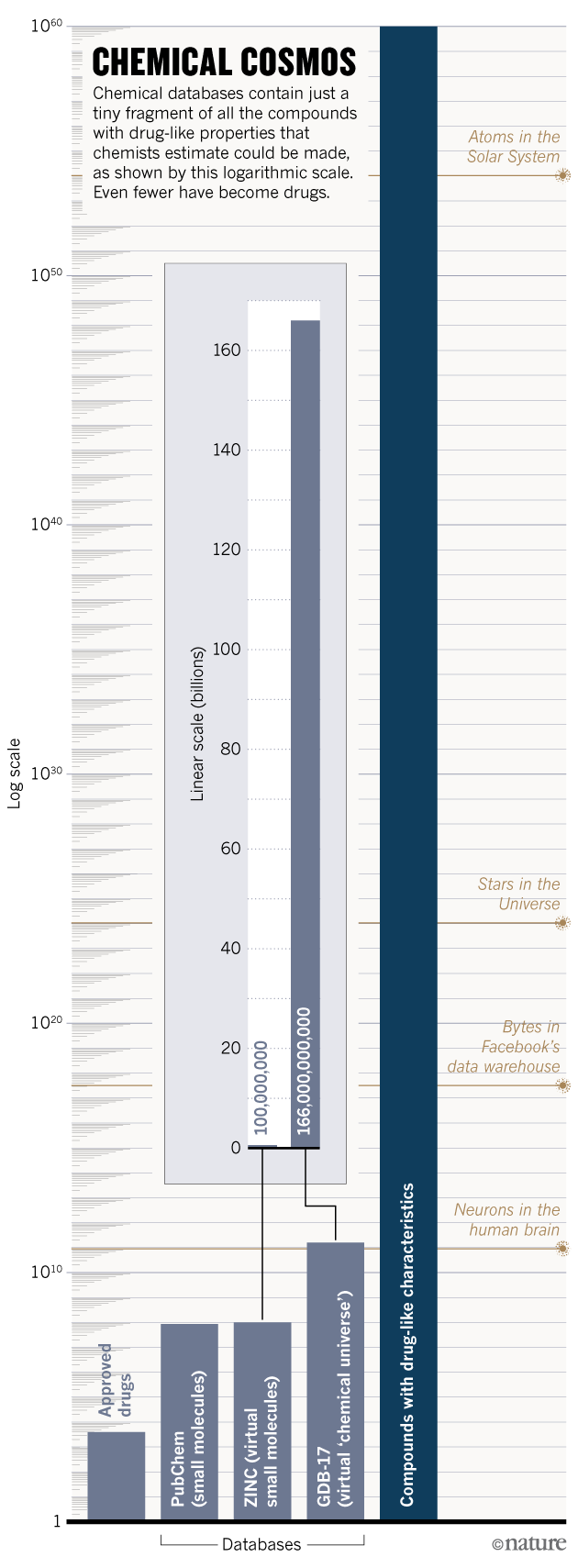
Exscientia es sólo uno de los numerosos grupos de la industria y el mundo académico que están recurriendo a las computadoras para explorar el asombroso y gran universo químico. Los químicos estiman que se podrían hacer 10^60 compuestos con características parecidas a las de un fármaco, es decir, moléculas más pequeñas que átomos hay en el Sistema Solar. La esperanza es que los algoritmos catalogarán, caracterizarán y compararán las propiedades de millones de compuestos para ayudar a los investigadores a encontrar rápidamente y de manera asequible los mejores candidatos para un objetivo. Los defensores argumentan que estas estrategias podrían hacer que los medicamentos sean más seguros, asegurar que menos medicamentos fallen en los ensayos clínicos y permitir el descubrimiento de nuevas clases de terapias. También podrían ayudar a abrir áreas de espacio químico que quedan inexploradas o supuestamente estériles.
Pero muchos químicos medicinales se muestran escépticos ante la exageración, no convencidos de que la inefable complejidad de la química pueda reducirse a simples líneas de código. Incluso los defensores de la IA reconocen que muchos intentos han fracasado: los compuestos generados por computadora pueden estar plagados de componentes difíciles de fabricar, como los anillos de 3 o 4 átomos, e infestados con grupos reactivos que activan las alarmas de seguridad. «La ejecución de algunos enfoques computacionales puede sufrir mucho cuando los investigadores simplemente no conocen el campo», dice van Hoorn. «Los compuestos que se les ocurren son simplemente ridículos», pero afirma que un toque humano experto podría domar a estos diseñadores digitales demasiado entusiastas. «Creo que algunas de estas ideas podrían funcionar si los informáticos colaboraran con gente que realmente respira química.»
Exploración espacial
Para navegar por el universo químico, ayuda tener un mapa. En 2001, el químico Jean-Louis Reymond, de la Universidad de Berna (Suiza), comenzó a utilizar ordenadores para trazar la mayor parte del espacio masivo posible. Dieciséis años después, ha acumulado la mayor base de datos de moléculas pequeñas del mundo, una gigantesca colección virtual de 166 mil millones de compuestos. La base de datos, llamada GDB-17, incluye todas las moléculas orgánicas químicamente factibles, compuestas de hasta 17 átomos, tantas como los ordenadores de Reymond puedan soportar. Sólo para que un ordenador pueda compilar una lista de los compuestos de la base de datos llevaría ahora más de 10 horas», dice Reymond.
Para dar sentido a esta plétora de posibles puntos de partida de medicamentos, Reymond ha ideado una forma de organizar su universo químico. Inspirándose en la tabla periódica, ha agrupado los compuestos en un espacio multidimensional en el que los compuestos vecinos tienen propiedades relacionadas. Las posiciones se asignan de acuerdo a 42 características, tales como cuántos átomos de carbono tiene cada compuesto.
Por cada medicamento que ha llegado al mercado, hay millones de compuestos que son químicamente casi idénticos a él – solo que tienen un hidrógeno extra aquí o doble enlace allí. Y algunos de estos funcionarán mejor que el medicamento aprobado. Los químicos no podrían concebir todas estas variaciones sin ayuda. «No hay manera de llegar a estos isómeros usando un bolígrafo y un pedazo de papel», dice Reymond.
Pero Reymond y su equipo pueden identificar «vecinos cercanos» terapéuticamente prometedores de drogas probadas mediante la búsqueda de similitudes entre los compuestos. Al utilizar un medicamento en particular como punto de partida, el equipo puede peinar los 166 mil millones de compuestos en la base de datos para obtener candidatos de seguimiento convincentes en sólo 3 minutos. En un experimento de prueba de principio, Reymond comenzó con una molécula conocida que une el receptor de acetilcolina nicotínica, un objetivo útil para trastornos que involucran el sistema nervioso o la función muscular, y compiló una lista corta de 344 compuestos relacionados. El equipo sintetizó tres, y descubrió que dos podrían activar el receptor de forma potente, y podrían ser útiles para tratar la atrofia muscular en el envejecimiento. El enfoque es como usar un mapa geológico para determinar dónde buscar oro, dice Reymond. «Necesitas alguna forma de elegir dónde vas a cavar», dice.
Un enfoque alternativo utiliza las computadoras para buscar oro en muchos lugares sin preocuparse demasiado por la ubicación inicial. En términos de búsqueda de medicamentos, esto significa que se deben buscar en sílice grandes bibliotecas químicas para encontrar pequeñas moléculas que se unen a una proteína dada. Primero, los investigadores tienen que tomar una instantánea de una proteína usando cristalografía de rayos X para determinar la forma de su sitio de unión. Luego, usando algoritmos de acoplamiento molecular, los químicos computacionales pueden escarbar a través de colecciones de compuestos para encontrar los mejores ajustes para cualquier sitio dado.
A medida que la potencia de computación ha explotado, las capacidades de estos algoritmos han mejorado. Los químicos de la Universidad de California en San Francisco, encabezados por Brian Shoichet, mostraron el potencial de este enfoque en 2016 en la búsqueda de una nueva clase de analgésicos. El equipo revisó más de tres millones de compuestos comercialmente disponibles para encontrar candidatos que activaran selectivamente la señalización del receptor de opiáceos para aliviar el dolor sin alterar la vía de señalización de detención de opiáceos estrechamente relacionada, que se cree que está asociada con los efectos secundarios de los opiáceos, incluyendo una menor frecuencia respiratoria y estreñimiento. Los investigadores rápidamente redujeron una biblioteca masiva de compuestos a sólo 23 compuestos altamente clasificados para el seguimiento.
En el tubo de ensayo, siete de los candidatos tuvieron la actividad deseada. El desarrollo posterior convirtió a uno de estos en PZM21, un compuesto que actúa sobre el receptor de los opiáceos sin activar la beta-arrestina. La firma biotecnológica Epiodyne, con sede en San Francisco, California, y cofundada por Shoichet, está ahora tratando de desarrollar un analgésico más seguro basado en los hallazgos. Shoichet planea utilizar el mismo enfoque para encontrar compuestos que modulen otros receptores acoplados a la proteína G (GPCR), una familia de proteínas que representa aproximadamente el 40% de los objetivos de medicamentos.
Su equipo también está llevando a cabo experimentos similares con una nebulosa virtual de 100 millones de compuestos que nunca se han hecho antes, pero que debería ser fácil de sintetizar. Los desarrolladores de medicamentos industriales también están probando este enfoque: la firma biotecnológica Nimbus Therapeutics, con sede en Cambridge, Massachusetts, incorpora en sus pantallas de acoplamiento compuestos virtuales con características de sustancias químicas naturales que normalmente tienen que ser obtenidas laboriosamente de entornos naturales como el suelo. El jurado todavía no está claro si esto conducirá a los medicamentos, pero Don Nicholson, director ejecutivo de la compañía, dice que para al menos un programa de diseño de fármacos, «de aquí vienen todos nuestros éxitos».
Los resultados preliminares de este tipo de pruebas virtuales están sacudiendo uno de los supuestos básicos de Shoichet sobre el espacio químico: que sólo vale la pena mirar en regiones establecidas y ricas en medicamentos. Galaxias bien caracterizadas de moléculas están tan inundadas con compuestos biológicamente activos que algunos argumentan que es una pérdida de tiempo buscar en otro lugar. «A lo largo de mi carrera he creído esa línea de razonamiento. Tenía sentido, aunque no hubiera tanta evidencia que lo apoyara «, dice Shoichet. Pero los resultados inéditos de sus cribas de 100 millones de compuestos están avivando su interés en las regiones menos exploradas del espacio químico. «Empiezo a pensar que esas galaxias están llenas de oro.»
… …
Ampliar en: nature
![]()

Premio Nobel de Química 2013

A estas alturas del año, seguro que todos ustedes saben que la Real Academia de Ciencias de Suecia ha otorgado el Nobel de Química 2013 al investigador austríaco Martin Karplus, al sudafricano Michael Levitt y al israelí Arieh Warshel por el “desarrollo de modelos multiescala de sistemas químicos complejos”…. cosa que es muy posible que a alguno de ustedes les suene a chino.
La concesión a estos tres investigadores de la máxima distinción científica en el campo de la alquimia ha levantado mucha polémica, pero desde mi punto de vista es más que merecida ya que sus descubrimientos en el campo de la química computacional están siendo de vital importancia en el desarrollo de muchas otras áreas científicas, lo que está repercutiendo positivamente en el día a día de los ciudadanos.
En el blog SCIENTIA se trata «¿Puede el Premio Nobel de Química 2013 salvarte la vida?» del que se pueden extraer las siguientes conclusiones:
Por un lado el desarrollo de la química computacional está siendo de vital importancia para la predicción y desarrollo de infinidad de reacciones químicas de gran importancia por lo que la concesión del Premio Nobel a los padres de esta rama de la química está más que justificada.
Por otra parte hemos de tener claro que la química computacional y la química experimental forman un binomio en el cual cada uno de los componentes se nutre del otro. La química computacional necesita a la experimental para corroborar sus predicciones y la química experimental necesita a la computacional para optimizar su rendimiento.
Pero la conclusión más importante que yo personalmente extraigo es la siguiente. En este post les he mostrado como en un solo trabajo un grupo de investigadores emplea herramientas pertenecientes a campos tan “teóricamente” distintos como la química, la informática, la medicina, la bioquímica, la farmacología, las matemáticas… lo que me ha servido para ratificarme en algo que llevo pensando muchos tiempo: en los tiempos que corren parcelar áreas científicas tiene cada vez manos sentido. El trabajo multidisciplinar es el futuro del avance científico.
![]()
Premio Nobel Química 2013: Karplus, Levitt y Warshel por la bioquímica computacional

El austríaco Martin Karplus (Univ. Harvard, Cambridge, Massachusetts, EEUU), el sudafricano Michael Levitt (Facultad de Medicina de la Univ. Stanford, California, EEUU) y el israelí Arieh Warshel (Univ. del Sur de California, EEUU) son los ganadores del Premio Nobel de Química 2013 por “el desarrollo de modelos multiescala para sistemas químicos complejos.” La simulación por ordenador de la química cuántica de las macromoléculas (como las proteínas) y sus interacciones con los metabolitos (moléculas pequeñas) es imposible; el número de grados de libertad crece de forma exponencial con el tamaño. Por fortuna, basta simular la física cuántica de la reacción en el sitio activo (o centro de reacción), pudiendo usarse la mecánica clásica de Newton para simular las vibraciones del resto de la molécula, la llamada dinámica molecular. Este tipo de simulación multiescala fue introducida por Karplus, Warshel y Levitt entre 1972 y 1976. Desde entonces se considera la técnica numérica estándar para simular procesos bioquímicos en macromoléculas.
Anuncio del premio Nobel, información divulgativa [PDF], información técnica [PDF] y un artículo periodístico de Antonio Martínez Ron, “Nobel de Química 2013 para los científicos que facilitaron las simulaciones químicas por ordenador,” lainformacion.com, 9 Oct 2013.
Ampliar en: Francis (th)E mule Science’s News
![]()

autobus las palmas aeropuerto cetona de frambuesa